An Overview of Growth, Aging, Senescence, and Immortality in our HMEC Culture System
(last updated 12/2019)
Introduction
The goal of our studies since 1976 has been to understand the normal processes governing growth, aging, and senescence of HMEC, and how these normal processes are altered during immortal and malignant transformation. To address these goals, our lab has generated a large variety of HMEC types, ranging from primary organoid material and normal HMEC strains, to isogenic series of cultured cells at various stages of transformation (Chart 1). Examination of these cultures has elucidated the many significant differences between normal and abnormal HMEC, and produced a new, molecularly defined model of the senescence barriers encountered by cultured HMEC (Figure 1, Table 1). These senescence barriers function to suppress tumorigenesis, thus understanding how they are overcome as normal cells transform to immortality and malignancy can provide insight into the mechanism of human malignant progression in vivo, and into possible therapeutic interventions. Our HMEC culture system has been shown to accurately model many aspects of early stage breast carcinogenesis in vivo, and can serve as an experimentally tractable model to examine factors that influence human cellular aging and carcinogenesis. Notably, our studies highlight the importance of the (often overlooked) immortal transformation step in malignant progression. Our ability to achieve long-term growth of normal finite HMEC of multiple lineages makes possible extensive examination of normal HMEC biology, differentiation, and aging. We believe that understanding aberrant biological process requires prior knowledge of the normal finite state.
Early Stage Human Breast Carcinogenesis In Vivo
Our aim has been to develop a culture system that can correctly model the in vivo processes seen in normal HMEC, and the alterations observed during malignant transformation, in order to optimize the value of our work for human health. While large gaps in knowledge remain, much data has been gained in the past decades about human breast cancer progression, and we have used this information in both the design and assessment of our HMEC system. Some relevant points:
- The normal breast contains multiple epithelial types (stem, progenitors, myoepithelial, luminal) embedded within a complex stromal matrix; all these normal cells have a finite lifespan; no normal HMEC is immortal. Normal HMEC function as polarized cells.
- Studies have suggested that the cells of origin for most breast cancers are progenitors: luminal progenitors lead to basal type cancers (1-3) and probably also luminal cancers, whereas basal cells may be the origin of claudin low and metaplastic cancers (4).
- Some early abnormalities are associated with increased cancer risk, e.g., ~2-fold for hyperplasias, ~5-fold for atypical hyperplasia (5,6). These lesions are common (600-800,000/year), and their prevalence increases with age (7), so the step from normal to hyperplasia does not appear stringently limiting.
- Many early epithelial cell abnormalities are associated with errors in the RB pathway (8-11). Mutations in PIK3CA can also be found at this stage (12,13).
- Pre-malignant DCIS is where critically short telomeres, genomic instability, and telomerase activity (immortalization) are first observed (14-18). Notably, many studies have shown that the molecular and genomic properties of cells in high-grade DCIS closely resemble what is seen in the cognate primary cancer, rather than normal finite HMEC (13,19-24).
- There is ~30% rate of progression from DCIS to primary cancer (25), so the step from pre-malignant to invasive malignant does not appear to be stringently limiting.
- Breast cancer lines/tissue display very short telomeres (~4 kb mean TRF, shorter than viable normal finite cells) and modest telomerase activity (26,27).
We recognize that an accurate transformation model would need to address the 3D, cell-cell, and cell-matrix interactions occurring in vivo. Still, it is important that the properties of the cells employed in 3D formats resemble what is found in vivo. We emphasize this point, as we view much current work involving HMEC culture and transformation as utilizing cells that obviously do not model the in vivo situation (e.g., see below on the commonly used commercially available highly aberrant post-stasis post-selection HMEC being sold and used as “normal”, on the non-cancer-like immortalization achieved using hTERT, and on calling non-malignant immortally transformed HMEC “normal” or “untransformed”).
Model
We postulate that cultured finite lifespan HMEC encounter at least two mechanistically distinct barriers to indefinite proliferation, stasis (stress-associated senescence) and replicative senescence (telomere dysfunction due to telomere attrition). Finite HMEC are also vulnerable to oncogene-induced senescence (OIS). Errors are needed to bypass or overcome the stasis and replicative senescence barriers. Once cells become immortal (telomerase expressing) they are no longer vulnerable to OIS, and a gain-of-function oncogenic error can be sufficient to confer malignant properties.
Fig.1. Model of senescence barriers encountered by cultured HMEC
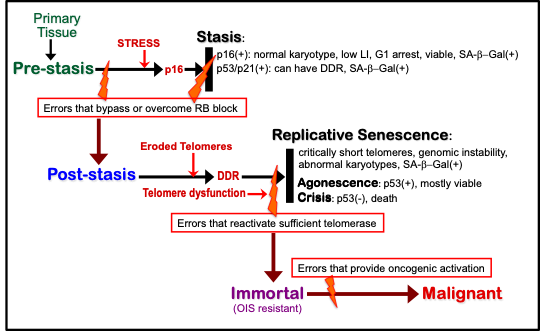
Table.1. Molecular properties of cultured HMEC at senescence barriers
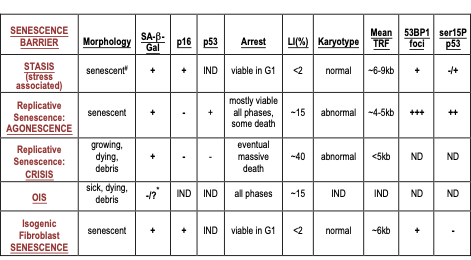
IND: senescence barrier independent of that property; ND: not determined
#: morphology of HMEC arrested at stasis in MCDB170 shows abundant stress fibers
*: negative in pre-stasis HMEC; could not be determined in post-selection HMEC
The expression of 53BP1 foci and serine 15 phosphorylated p53 are markers of a DDR.
Stasis is a stress-associated barrier, mediated by the retinoblastoma (RB) pathway, that is independent of telomere length and extent of replication. Stasis can be bypassed or overcome in cultured HMEC by multiple types of single alterations (genetic and/or epigenetic) in pathways governing RB, and does not require loss of p53 function (28-32). Overcoming stasis may correlate with hyperplasia/atypical hyperplasia in vivo, which commonly display clonal growth and errors in the RB pathway (e.g., loss of p16 expression, mutated RB, overexpressed cyclin D1) (8,9,11,30,33). Gross genomic aberrations are not common at this stage in vivo (14), and are not associated with overcoming stasis in vitro (34,35).
The onset of stasis in cultured HMEC correlates with increased expression of p16, but not p21 (30,34-36). The number of population doublings (PD) achieved prior to stasis depends upon culture conditions; we have observed a range of ~10-60 PD (28,35-38). Molecular correlates that can identify stasis, in addition to p16 expression, include arrest in G1, low labeling index (LI), non-critically short telomeres and normal karyotypes (34-36). These parameters are consistent with an RB-mediated arrest and the absence of a significant DNA damage response (DDR). Cells at stasis express senescence-associated β-galactosidase (SA-β-Gal) activity and a senescent morphology.
We postulate that stasis can also be enforced by p53-dependent p21 in response to DNA damaging stresses such as oxidative damage or radiation. Although neither cultured HMEC or their isogenic mammary fibroblasts express p21 at the onset of stasis, many other epithelial and fibroblast cell types may be more vulnerable to DNA damage inducing stresses in culture. They may express p21, and show greater evidence of a DDR at stasis, including foci which preferentially form at the telomeres due to their high GC content and low repair capacity (39,40). However, such telomeric damage is distinct from critically shortened telomeres at replicative senescence due to lack of telomerase. HMEC in vivo may also experience p53-inducing stresses. The p53-dependent type of stasis arrest does not require critically short telomeres or genomic instability, and inactivation of p53 (or p21) function may facilitate overcoming this arrest (41,42). Reactivation of telomerase is neither necessary nor sufficient to overcome stasis. NOTE: The p16(+) alone stasis seen in cultured HMEC is not associated with a significant DDR. A DDR is not an obligate consequence of stress-induced senescence. I postulate that microenvironmental and structural stresses (deviations from appropriate homeostasis) may contribute to p16 induction, without being genotoxic stressors. This may be relevant to the in vivo association of p16 and aging.
Replicative senescence occurs in post-stasis HMEC (cells that have bypassed or overcome stasis) due to ongoing proliferation producing progressively shortened telomeres, in the absence of sufficient telomerase activity. When telomeres become critically short (mean TRF ≤ 5 kb), telomere associations, genomic instability, and a DDR are elicited. Where wild-type p53 is present, most cells show a viable arrest; this barrier has been termed agonescence (34,36,43). Karyotypic analysis of HMEC at agonescence has shown that virtually all metaphases exhibit gross chromosomal abnormalities, predominantly telomere associations (34). This result is not consistent with a hypothesis that a p53-dependent senescence arrest due to telomere attrition occurs as soon as one uncapped telomere is present (44,45). When p53 is non-functional a viable arrest is not possible, and crisis-associated massive cell death occurs (36). Since many post-stasis HMEC lack p16 function, p16 is not necessary for replicative senescence.
The barrier due to telomere attrition can be overcome by the expression of sufficient telomerase to maintain stable telomere lengths. Overcoming replicative senescence may correlate with high-grade DCIS in vivo, which commonly display short telomeres, genomic instability, and telomerase reactivation (14-18). That most DCIS contain critically shortened telomeres indicates that the precursor cells did not express sufficient telomerase for telomere maintenance.
In HMEC, agonescence can be distinguished from stasis by the presence of critically short telomeres and genomic instability, higher LI (~15%), arrest at all phases of the cell cycle, and presence of a DDR (Table 1). HMEC at agonescence as well as at stasis display a senescent morphology and SA-β-Gal, so these properties do not readily distinguishable between these two molecularly distinct senescence barriers. Crisis can be distinguished from agonescence in HMEC by a higher LI (~40%) and the absence of a viable arrest. Since most human epithelial and fibroblast cells induced to transform to immortality in culture had inactivation of p53 function to facilitate overcoming stasis (e.g., using viral oncogenes or inhibitors of p53 function), only crisis was observed in such cultures at replicative senescence.
Cultured finite lifespan HMEC are vulnerable to oncogene-induced senescence (OIS) (46,47). HMEC that have attained immortality via reactivation of endogenous telomerase are no longer vulnerable to OIS, and show gain of malignancy-associated properties when exposed to oncogenes such as Raf-1, Ras, Insulin R, or ErbB2 (46,48-50). HMEC immortalized by exogenous hTERT transduction appear initially sensitive to OIS (46). The molecular correlates of OIS in HMEC differ from those seen in cells at stasis or telomere dysfunction (Table 1). OIS in HMEC does not require p16 or p53 function, and is independent of telomere length; its molecular properties are consistent with a DDR (46,47).
Additionally, finite HMEC may cease growth due to terminal differentiation. We observe terminal luminal cells (visible as flat non-refractile cells in patches with smooth edges), but have not investigated this process.
NOTE: We also draw attention to Table 1 and the similarity between the properties of senescent human mammary fibroblasts and HMEC stasis, but not HMEC replicative senescence. We believe a careful review of the literature of human fibroblast cells at senescence does not support the general assumption that this (Hayflick limit) is due to critically short telomeres inducing replicative senescence, as in most cases where measured, the telomeres have, as above, a mean TRF of ~6 kb - not critically short - and there is not evidence of significant telomere-associations. In my opinion, a confusion between replicative senescence (due to critically short telomeres) and stasis (which can involve telomere damage and shortening) has handicapped the senescence field for decades. Some of this may be due to attributing the presence of activated p53, a DDR, and telomeric foci in stasis to replicative senescence. Much of the literature may make more sense when what is referred to as replicative senescence is seen as stasis. Further, much of the molecular expression in senescent fibroblasts is not recapitulated in senescent HMEC (35), so the common use of just fibroblasts for studies on senescence may not provide accurate information about senescent epithelial cells.
Finite Lifespan HMEC: pre-stasis and post-stasis
HMEC derived from reduction mammoplasties, milk, benign tumors, and non-tumor mastectomy tissues have been grown in either serum-containing (MM, M85, M87A) or serum-free (MCDB 170) media (Chart 1)(28,35,37,38,51-53). Depending upon the media and culture conditions, active proliferation has ceased after ~10-60 PD (Figure 2). In media that support fewer PD, e.g., MCDB170, levels of p16 expression increase early; virtually all cells express p16 at stasis in all media used (30,35). The molecular profile of the HMEC at stasis is similar regardless of their PD potential or growth media (Table 1), with one noticeable difference. HMEC grown in serum-containing media have a typical senescent morphology of large flat vacuolated cells, whereas HMEC that had been grown in serum-free MCDB170 exhibit a more elongated morphology showing abundant stress fibers (28,35,52). We believe this difference is due to the serum-free medium being more stressful for cultured HMEC, consistent with the early rise of p16 and the low PD potential of HMEC initiated in MCDB170. Similar results are seen in commercial serum-free MEGM (Lonza) and M171 (Thermo Fisher), which are based on our original MCDB170 medium (28,30,53). This difference in morphology may have led other investigators to consider this stasis arrest distinct, and refer to it as “M0” (54,55).
Pre-stasis in M87A: Our most recent medium, M87A, supports long-term growth of normal pre-stasis HMEC (Figures 2,3) (35,53). The pre-stasis HMEC we now distribute were grown in this medium. Populations contain a mixture of cells with markers of myoepithelial, luminal, and progenitor lineages; later passage cultures show fewer luminal cells (56,57). Cells with luminal markers may also cease proliferation due to terminal differentiation rather than p16(+) stasis. Senescent cells remain genomically stable. We have examined gene transcript profiles, global promoter methylation, and DDRs as a function of passage from several individual’s HMEC (32,35). As expected, gene expression changes significantly with passage, while no obvious differences were seen for promoter methylation. Some interindividual differences could be detected in gene expression and extent of DDRs. Previous studies have shown interindividual differences in carcinogen metabolism (58), leading us to recommend that at least 2 individuals be examined to determine normal HMEC properties. We have also observed differences in lineage markers and differentiation correlated with the age of the specimen donor (56,57,59-61). With increasing age, HMEC from 4th passage pre-stasis strains and from uncultured dissociated organoids showed a decline of myoepithelial cells, and an increase of luminal cells that exhibited molecular features usually ascribed to myoepithelial cells (increased expression of integrin a6 and keratin (K)14). The proportion of c-Kit expressing cells (putative progenitors) also increased with age, and exhibited impaired microenvironment-directed differentiation and lineage specificity, consistent with alterations in Hippo pathway activation. These data suggest that the observed age-associated increase in luminal breast cancer could be connected to changes that occur normally with aging in the human breast. Myoepithelial cells are thought to be tumor-suppressive and progenitors are putative etiological roots of some breast cancers. Thus, during the aging process, the potential target cell population may increase while there is a simultaneous decrease in the cells thought to suppress tumorigenic activity.
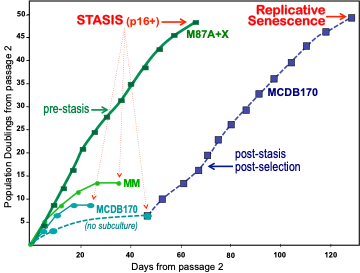
Figure 2. Population doubling potential of pre-stasis HMEC in different media. Primary cultures from reduction mammoplasty specimen 184 were initiated from organoids in different media. The number of PD in primary culture cannot be accurately determined; growth is shown starting from passage 2. All proliferation stopped in HMEC grown in serum containing media (MM and M87A with oxytocin (X)) after 10-50 PD beyond passage 2. The extensive proliferative potential in M87A+X supports generation of large batches of early passage pre-stasis HMEC from individual donors. HMEC initiated in serum-free MCDB 170 (commercial MEGM) show rapid induction of p16 and cessation of growth. When cultures are allowed to sit without subculture for 2-3 weeks, post-selection post-stasis HMEC emerge and maintain growth to replicative senescence. If cultures are repeatedly subcultured, fewer to no post-selection cells may emerge. All HMEC, regardless of growth medium, expressed p16 at stasis.
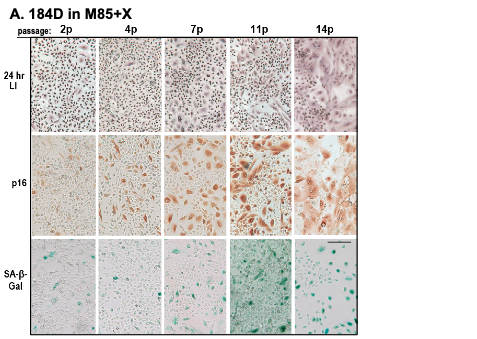
Figure 3. Characterization of pre-stasis HMEC grown in M85 with oxytocin. A. Expression of markers associated with proliferation (LI) and senescence (p16, SA-β-Gal) in pre-stasis 184D HMEC with increasing passage; stasis was at passage 15. Note the reciprocal relationship between the small cells with a positive LI, and the larger, often vacuolated cells (senescent morphology) that are positive for p16 and SA-β-Gal, and negative for LI. Size marker = 200 microns.
Pre-stasis and post-stasis in MM: We have cultured pre-stasis HMEC from over 150 individuals in serum-containing media (MM and M85/87A) and have not observed even a single instance of a cell spontaneously overcoming the stasis barrier. However, early experiments that exposed primary cultures of specimen 184 HMEC grown in MM to the chemical carcinogen benzo(a)pyrene (BaP) resulted in the emergence of HMEC colonies that maintained growth after the bulk of the cultures ceased proliferation at stasis (29,62). These BaP post-stasis populations (originally called Extended Life), ceased growth after an additional 10-40 PD, with the exceptional of very rare cells that became immortal cell lines (see below and Chart 1). All three BaP post-stasis cultures that have been examined showed loss of p16 expression, associated either with mutation or promoter silencing (11,30). Whole exome analysis (63) indicates that these cultures contain ~100-200 BaP-induced point mutations, the majority consistent with the known mutation spectrum of BaP (64). We have very limited quantities of BaP cultures available for distribution on a collaborative basis. A different post-stasis type emerged from pre-stasis cultures exposed to the genetic suppressor element GSE22 (65), which inhibits p53 function (36). Although p53 inactivation does not bypass/overcome stasis in these cultured HMEC, rare clonal escape from stasis was observed (presumably due to induced instability), leading to the GSE22-post-stasis cultures (Chart 1).
Pre-stasis and post-stasis in MCDB170: When the HMEC are cultured in the highly stressful serum-free MCDB170 medium (commercial equivalents: Lonza MEGM; Thermo Fisher M171), a small number of cells are able to overcome stasis in the absence of additional oncogenic exposures (28) (Figure 2). These post-stasis cells are highly aberrant: 1) they show methylation of the p16 promoter and absence of p16 expression, as well as nearly 200 other mostly cancer-associated changes in promoter methylation (in contrast to the BaP post-stasis cultures, which display only ~10 changes) (30,32); 2) their gene transcription varies significantly from isogenic normal pre-stasis HMEC (35,66); 3) they show rapid loss of lineage heterogeneity (53); 4) they have metaplastic properties and may be on a pathway to metaplastic cancer (67).
We originally called the emergence of these post-stasis cells “selection” and this class of post-stasis HMEC “post-selection”. We now recognize that selection (what other labs later termed “M0”) is a stasis arrest. Although the pre-stasis populations may be heterogeneous with respect to a cell’s ease in silencing p16 to become post-selection (68), we believe the post-selection cells are induced by growth in the stressful (oncogenic) serum-free MCDB170 medium, i.e., post-stasis cells are not present in the starting normal pre-stasis cultures. This is based on the total absence in over 40 years of our work of any post-stasis cell emerging from normal pre-stasis HMEC grown in any of our serum-containing media, as well as the absence or reduction of post-selection HMEC emerging from pre-stasis HMEC grown in MCDB170 when there are small changes in media composition (e.g., absence of a cAMP stimulator) or methodology (e.g., sub-culturing cells approaching stasis/selection rather than waiting 2-3 weeks at stasis without subculture for the post-selection cells to emerge; we presume the induction of the p16(-) cells is occurring during this time when no cell divisions are observed). It is likely that post-stasis cells exist in some breast tissues although the post-stasis type is not identified. Rare p16(-) HMEC have been seen in apparently normal breast tissues in vivo (68). These rare cells have been called vHMEC but the nature of the error(s) leading to the silencing of p16 in vHMEC in vivo is not known. The term vHMEC has also been used by others to refer to p16(-) post-stasis cells in culture that are specifically post-selection.
NOTE: These highly aberrant post-selection post-stasis HMEC are what is sold commercially as normal primary HMEC, e.g., Lonza CC-2551 and Thermo Fisher A10565. I’ve had several puzzled scientists call me after purchasing these cells to describe what they saw as non-normal behavior – because these cells are not normal! - and I’ve been unable to effect any change in this commercial situation. My speculative hypothesis, based on working with Dick Ham’s lab on developing MCDB 170 (28), is that their serum-free medias may be stressful in mass culture because they were optimized using clonal assays (they support clonal growth). They may not be best for mass culture, and the prevalence of these serum-free media for culturing human epithelial cells could be introducing misrepresentations of normal behavior.
Post-selection post-stasis p16(-) HMEC grow actively for an additional ~30-70 PD, depending on the individual. They express wild-type p53 that is present in a stable form (36,69,70). As they near agonescence, they exhibit a senescent morphology, SA-β-Gal, a DDR, and genomic instability (34,36). If p53 function is inactivated (e.g., using GSE22) cells continue to proliferate for an additional ~2-4 passages, with increasing evidence of cell death and debris (i.e., crisis) (Figure 4)(36). The telomere dysfunction barrier is extremely stringent in post-selection post-stasis HMEC. We have never seen any unperturbed cell at agonescence spontaneously immortalize. We have also never seen any immortalization at crisis in post-selection HMEC, but rare immortalization at crisis using DN-p53 constructs has been reported by others (71,72). This stringency is due, in part, to the molecular nature of this barrier; cells that fail to maintain a G1 or G2 arrest with critically short telomeres will eventually die or become non-proliferative as a consequence of the genomic instability and mitotic catastrophes. Overcoming replicative senescence requires induction of sufficient telomerase. However, in post-selection post-stasis cells (see more below), it appears that more than one error is needed to reactivate telomerase. The probability of this occurring in one p53(+) cell during telomere dysfunction is exceedingly low, accounting for the lack of immortalization in the post-selection cultures at agonescence (as distinct from our other post-stasis types, which exhibit rare clonal immortalization).
We have large supplies of post-selection HMEC available for distribution from women of various ages. It’s important to recognize that these cells are not normal, and acquire genomic instability as they are propagated in culture. In the past, due to the inability to attain long-term culture of normal pre-stasis HMEC, we provided post-selection HMEC for studies on finite HMEC. Since it is now possible to grow large quantities of normal pre-stasis HMEC, we strongly recommend that studies aiming to understand normal HMEC behavior use HMEC that are normal, and not the aberrant post-selection HMEC (from us or commercial sources). For some experimental purposes, post-selection or other post-stasis HMEC may be preferable, e.g., examining the requirements for and mechanisms of overcoming the telomere dysfunction barrier, or assaying cells at different stages in progression.
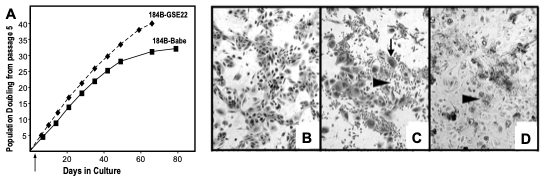
Figure 4. Growth and morphology of post-stasis post-selection 184 with and without functional p53. 184B HMEC were transduced with GSE22-containing or control (Babe) vectors at passage 5. (A) growth curves of 184B-Babe and 184B-GSE22. Note the additional PD in the cultures lacking functional p53. We believe growth rates are similar ± p53, but the absence of p53-mediated growth inhibition allows more cells to continue to proliferate to crisis, leading to apparent faster growth of the population as cells near telomere dysfunction. (B) 184B-Babe at agonescence, 2 months after plating at passage 15, contains mostly large, flat cells with some vacuolization; the cell population can retain this morphology and viability for over a year. (C) 184-GSE22, two weeks after plating at passage 15, shows areas of small proliferating cells and many very large flat cells (arrows). (D) 184B-GSE22, four months after plating at passage 15, shows mostly large multi-nucleated, vacuolated cells and abundant cell debris. All photographs are at the same magnification. (36)
Post-stasis in M85 or M87A: More recently, we have generated additional types of post-stasis HMEC using shRNA to p16 (p16sh) and transduction of a cyclin D1/CDK2 construct (D1) (Chart 1) (53,73). As expected, in early passage HMEC grown in M85 or M87A, direct inhibition of p16 using p16sh, or transduction of D1, led to widespread bypass of stasis. However, bypass with p16sh was (visually) greater in young vs older women’s HMEC, whereas for D1 bypass was greater in old vs young. p16sh and D1-post-stasis HMEC show few differences in their pattern of promoter methylation and/or gene transcripts compared to their precursor pre-stasis cultures (30,32,74). They grow for an additional ~20-40 PD until agonescence (Figure 5). Unlike post-selection post-stasis, and similar to BaP post-stasis cells, p16sh post-stasis HMEC have generated rare clonal immortal lines during the period of genomic instability at agonescence. This difference (see more below) is presumably due to the ability of just one error to immortalize these cultures (53,73). We can provide limited amounts of the p16sh post-stasis HMEC for specific requests. We’ve not extensively examined the D1 post-stasis; they show more PD than the p16sh before agonescence. No immortal lines have emerged from reduction mammoplasty derived HMEC, but immortal lines have arisen in D1-transduced milk derived HMEC.
I want to add a few comments about nomenclature since this issue has frequently come up in discussions. It’s my general experience in science that functionally distinct molecules or molecular processes are given distinct names. Confusion could result if there were not distinct names for different, though closely related family members (e.g., growth factors and their receptors) or related mechanisms (e.g., apoptosis, anoikis, mitotic catastrophe). One of our overall goals is to try to model the many different in vivo pathways a normal cell can take to become malignant. Such information may assist individualized clinical interventions. Our data thus far indicate that molecular properties differ among different pathways, starting with early stage carcinogenesis. Pre-stasis HMEC differ from post-stasis HMEC, although both are finite, and as discussed above, there are significant differences among the various post-stasis types. Since these different post-stasis cultures are functionally different, we have given them distinct names. Similarly, since agonescence is molecularly and morphologically distinct from crisis, although both result from telomere attrition, we believe it important that there be distinct names. Confusion may also arise if similar mechanisms are given distinct names, e.g., we view what we are defining as stasis as having also been referred to as M0, M1, MINT, M1.5, premature senescence, replicative senescence, and culture shock. Telomere dysfunction due to telomere attrition (replicative senescence displaying as agonescence or crisis) has been called replicative senescence, crisis, M2, and M1. Indeed, it is this situation that has prompted our efforts to generate molecularly defined nomenclature for the senescence barriers. I encourage everyone to employ the molecularly defined nomenclature we have presented here for our HMEC culture system.
Immortally Transformed Cell Lines
Introduction: The replicative senescence barrier can be overcome or bypassed by the expression of sufficient telomerase to maintain stable telomere lengths. However, unlike bypassing an arrest based upon keeping an active RB (e.g., abrogating the p16 expression that enforces stasis), the widespread chromosomal derangements caused by telomere dysfunction are not reversible. Consequently, in cultured HMEC, overcoming replicative senescence differs from bypassing/overcoming stasis in that the escaped immortal cells will contain the genomic abnormalities accumulated to that point, and may retain some degree of genomic instability (14,31,53,73,75). Our studies lead us to hypothesize that the inherent genomic instability consequent to telomere attrition can give rise to the errors permissive for telomerase reactivation, as well as many of the “passenger” errors seen in breast and other carcinomas, and that the generation of breakage-fusion-bridge (BFB) cycles prior to immortalization may underlie some of cancer-associated genomic instability (36,38,53,73). Thus many errors that can contribute to the ultimate cancer cell phenotype, including level of aggressiveness, may arise prior to immortalization and malignancy. This hypothesis is consistent with the in vivo situation as numerous publications indicate that many properties of invasive tumors are already present in their pre-invasive DCIS lesions, such as tumor markers, gene expression profiles, gene methylation, PIK3CA mutations, and genomic errors (13,19-24).
Importantly, our studies on immortalization support the critical role immortalization plays in human (but not murine) malignant progression, and it’s potential as a valuable therapeutic target. Studying the HMEC immortalization process as it occurs during cancer-associated immortalization in vivo has been difficult because it has been difficult to achieve immortalization in vitro using pathological relevant agents (i.e., not ectopic hTERT or viral oncogenes). Reported immortal lines have been rare clonal events (29,31,36,71-73,76-78). This likely reflects the fact that large long-lived animals such as humans have evolved mechanisms for stringent repression of telomerase in normal adult non-stem cells, presumably for tumor suppression. In contrast, cells from small short-lived animals such as mice do not show such stringent telomerase repression, and, lacking the replicative senescence barrier, readily immortalize once they overcome stasis (79-85). We believe that immortalization with telomerase reactivation is a rate-limiting step in human epithelial carcinogenesis, similar to the in vivo transition to high-grade DCIS, and thus great caution should be exercised in extrapolating mechanisms of rodent malignant progression to humans, as small rodents do not model the human tumorigenesis process because they lack a stringent barrier to immortalization. One of the goals of our long-term program in developing an HMEC model system of transformation has been to make available experimentally tractable human cells for examination of this crucial step in human malignant progression, since it cannot be accurately modeled in mice. As described further below, our initial work generated rare clonally immortalized lines containing multiple genomic errors, making it difficult to examine the immortalization process. More recent work has shown that efficient non-clonal immortalization is possible when the two main senescence barriers (stasis and replicative senescence) are directly targeted; resulting immortalized lines display normal karyotypes. These studies should facilitate experimental examination of the immortalization process in HMEC.
Clonal immortal lines: We have generated a variety of immortally transformed lines using various oncogenic agents (see: Chart 1 and Cell Types Generated) (29,31,32,36,46,53,62,73,76,86-89). Most of these lines were derived from post-stasis cultures, although in a few instances (involving hTERT or c-Myc transduction) lines emerged from perturbations of pre-stasis populations. Our first immortal lines were obtained from the BaP post-stasis cultures, 184Aa, 184Bd, and 184Be (29,31,62,63,86). Extremely rare clonal immortal lines have appeared at agonescence (184A1, 184AA4, 184AA8, 184B5, 184BE1, 184BE2). The BaP post-stasis cultures harbor small mutations from their BaP exposure, as well as loss of p16 expression (63). Large genomic errors such as copy-number variations (CNV) were seen in examined immortal lines but not the precursor BaP post-stasis cultures (63,75). Presumably, the genomic instability at agonescence produced the CNV and the rare errors that allowed telomerase reactivation and immortalization. More frequent but still rare clonal lines appeared at agonescence following transduction of the breast cancer–associated oncogene ZNF217 into the 184Aa population (184AaZN1-3 (76)). More frequent immortal clonal outgrowths at crisis were seen when p53 was inactivated in 184Aa using GSE22 (184AaGS1-2). All examined (karyology and/or aCGH) clonal lines exhibited numerous gross genomic errors. Uniform immortalization was obtained following transduction of c-Myc into three different BaP post-stasis cultures (184AaMY1-5, 184BeMY, 184CeMY (73), indicating that one error is sufficient to immortalize these BaP post-stasis cultures.
No post-selection post-stasis HMEC has been observed to spontaneously immortalize, presumably due to the need for two errors for immortalization. Rare immortal lines have appeared at agonescence following overexpression of the breast cancer associated oncogene ZNF217 (184ZN4-7) (73,76). Transduction of c-Myc alone produced 1 clonal line in 10 independent experiments (184SMY1). We hypothesize that rare errors generated by the genomic instability at agonescence may complement ZNF217 or c-Myc to allow telomerase reactivation. Overexpression of both c-Myc and ZNF217 in post-selection HMEC was able to produce clonal immortal lines in repeat experiments (184ZNMY1-4, unpublished); some of these clonal lines immortalized early, prior to agonescence.
Non-clonal immortal lines: More recently, we tested our hypothesis about the two main senescence barriers by targeting these barriers in early passage pre-stasis cells grown in M85 or M87A (53,73). If the genomic errors seen in clonal immortalized cells (in vitro and in vivo cancer) were required to bypass/overcome these barriers, but not needed per se, then directly targeting these barriers should produce non-clonal immortalized lines lacking gross genomic errors. That is indeed what we observed, using primary HMEC from 8 different specimen donors ranging in age from 19 to 91 (Figure 5). The stasis barrier was targeting using p16sh or D1/CDK2, and the replicative senescence barrier was targeted by transduction of c-Myc. Efficient immortalization has been seen with all specimens in all experiments. Karyotypes obtained from 6 different non-clonal immortalized lines (184Dp16sMY, 240Lp16sMY, 240LD1, 122Lp16sMY, 122LD1, 805Pp16sMY) were all normal diploid at early passage. From visual observation, there could be <100% transition to post-stasis using either p16sh or D1 alone, while the transition to immortality from the post-stasis cultures appeared close to 100%. c-Myc transduction of the BaP post-stasis cultures also appeared to give close to 100% immortalization efficiency. This transduction was done in 184Aa and 184Be after the start of telomere dysfunction, so the immortalized lines do not exhibit a normal karyotype, while 184Ce, transduced earlier, generated 184CeMY with a normal karyotype (though with many BaP-induced point mutations). The ability of the p16sh, D1, and BaP post-stasis cells to be immortalized by one error (i.e., c-Myc) likely accounts for the rare “spontaneous” clonal immortalization seen in these cultures at agonescence (184Fp16s, 184Dp16s1-2, 240Lp16s1-3, 805Pp16s, 254MKD1, 256MKD1 and the BaP-exposed lines). Amplification at the c-Myc region was observed in some of these clonal lines. We do not know why the post-selection post-stasis cells differ in this respect. We hypothesize that their prior experience of high stress may play a role. Transduction of c-Myc alone into pre-stasis HMEC in low stress-media gave rare clonal escape from stasis; these post-stasis cells with transduced c-Myc maintained growth to became clonal immortal lines (184FMY2, 184DMY3, 240LMY, 122LMY). All clonal lines examined showed multiple CNV by aCGH.
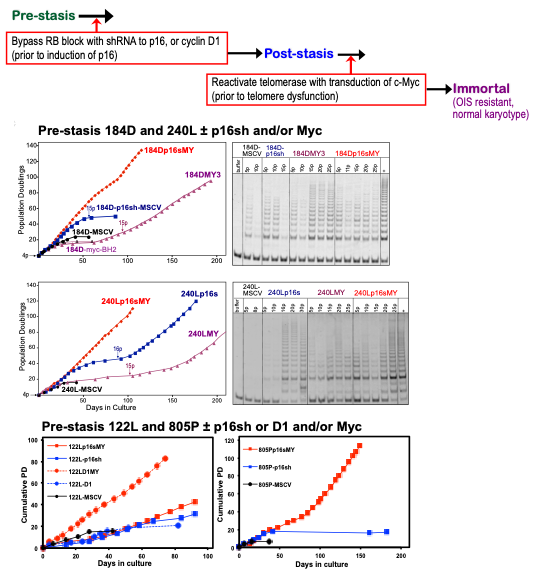
Figure 5. Effect of c-Myc on post-stasis HMEC growth and TRAP activity. Top panel: schematic representation of protocol to directly target senescence barriers to achieve non-clonal immortalization. Middle panel: Pre-stasis 184D and 240L HMEC grown in M87A+CT+X were transduced at 3p with a p16sh-expressing retrovirus (MSCV, blue) or empty vector (black). At 4p cultures ±p16sh were transduced with c-Myc (BH2)(red +p16sh; purple -p16sh). c-Myc-transduced p16sh post-stasis HMEC maintained active growth indefinitely, associated with increased TRAP activity. The continuous exponential growth following c-Myc transduction of the 4p p16sh-post-stasis populations reflects the observed non-clonal immortalization. Cells transduced with p16sh alone bypassed stasis and ceased net growth at agonescence, with rare clonal immortalization at agonescence. Cells transduced with c-Myc alone ceased growth at stasis, with rare clonal escape from stasis leading to immortalized lines. Control cultures transduced with empty vectors ceased growth at stasis. In some TRAP assays, heat-treated controls (+) were run next to unheated (-) samples. Positive TRAP control samples are indicted by “+”. E. Bottom panel: Pre-stasis 122L and 805P HMEC grown in M87A+CT+X were transduced at 3 or 4p respectively with a p16sh-expressing retrovirus (MSCV, blue), D1/CDK2 vector (blue dotted line) or empty vector (black). At 4p or 5p cultures ±p16sh or D1 were transduced with c-Myc (BH2)(red +p16sh; red dotted +D1). c-Myc-transduced post-stasis HMEC maintained active growth indefinitely. Each point indicates a passage.
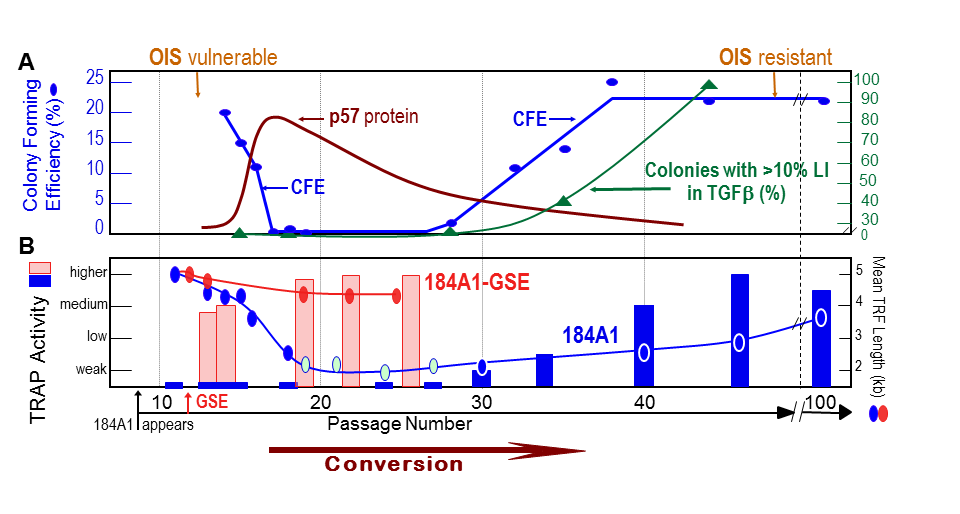
Figure 6. Conversion of newly immortal p53(+) HMEC lines is associated with changes in many key properties (A.B.) The p53(+) 184A1 line grown in MCDB170 initially exhibits low TRAP activity and ongoing telomere shortening until conversion occurs when the mean TRF declines to ~3 kb. Changes in growth capacity (CFE) and expression of p57 are also seen, and fully immortal lines gain the ability to maintain growth in the presence of TGFβ and resistance to OIS (31,46,76,86,90). When pre-conversion 184A1 is transduced with GSE22, there is a rapid increase in telomerase activity associated with stabilization of TRF length at ~4 kb (31).
Conversion to full immortality: Our studies have led to the discovery of a previously unreported step in HMEC immortalization, termed conversion, based on observing the need for multiple steps to attain a fully immortal potential (i.e., synthesizing sufficient telomerase to maintain stable telomere lengths) (31,36,38,46,73,76,86,88-90). Even after HMEC have acquired the errors allowing them to bypass/overcome both stasis and replicative senescence, and express hTERT, the resultant cells with indefinite proliferative potential still progress through further changes. This process has not been widely observed due to the difficulty in immortalizing human epithelial cells using pathologically relevant agents, and the absence of a significant immortalization barrier in small rodents.
Conversion has been most extensively studied in the immortal 184A1 line, which first appeared ~passage 8 in the 184Aa BaP post-stasis population and had a mean TRF value of ~5 kb when first examined at passage 11 (Figure 6). 184A1 (possibly due to its early immortalization relative to 184Aa replicative senescence) has only low level gross genomic errors, and no BFB; the lack of significant genomic instability facilitates examination of its immortalization process (63,75). We noted that cells that overcame replicative senescence gained the potential to express telomerase, but initially displayed little telomerase activity, and had ongoing telomere erosion with proliferation. When telomeres got extremely short (<3 kb), the conversion process ensued. When grown in the stressful MCDB170 medium, expression of the CKI p57Kip2 initially abruptly increased and then slowly declined, associated with initial slow-heterogeneous growth and then gradual re-attaining of uniform good growth. Telomerase activity gradually increased, and the faint very short telomeres seen during conversion gradually became stabilized with a mean TRF of ~3-7 kb. As telomerase activity increased, the immortal lines gradually developed the ability to maintain growth in the presence of TGFβ; this change is a direct consequence of the hTERT expression, as transduction of hTERT into post-selection HMEC confers the ability to maintain growth in TGFβ while producing uniform immortalization without evidence of conversion (88). A significant change that is associated with conversion and telomerase expression but not initial immortal potential is the loss of vulnerability to OIS (46). Since 184A1 first emerged in MM medium, and showed a much faster conversion in MM than in MCDB170, we knew that the speed of conversion could be influenced by external factors. More recent studies of pre-conversion 184A1 grown in M87A indicate that conversion can proceed with almost no slowdown in growth (another caution about using the stressful MCDB170 medium). Thus, at least partial inhibition of this critical, essential process can be mediated by currently unknown factors.
When we obtained immortal HMEC lines from 184Aa (grown in MCDB170) that lacked functional p53 (184AA2, 184AA3) we noted an initial brief slower growth phase and low TRAP activity, but they quickly increased telomerase activity and attained good uniform growth ± TGFβ and had no p57 expression. Their mean TRF length stabilized at ~4 kb and never declined to the very low levels seen in the p53(+) lines. The role of p53 in repressing telomerase activity in newly immortal lines grown in MCDB170 was then demonstrated by inactivating p53 (using GSE22) in pre-conversion 184A1 (Figure 6) (31). Endogenous telomerase activity was quickly expressed and mean TRF lengths stabilized; existing p57 expression was rapidly reduced. GSE22 transduction into the finite lifespan precursors of the immortal lines did not induce significant telomerase activity indicating that abrogation of p53 function alone is not sufficient for telomerase reactivation in post-stasis HMEC.
These results suggest that newly immortal lines have the potential to express telomerase, but expression is low until undergoing the conversion process (unpublished data have indicated that newly immortal p53(+) lines express low telomerase activity which can be inhibited). Fully immortal lines (p53+ and p53-) express short stable telomeres (mean TRF ~4 kb), resistance to OIS, and hundreds of promoter methylation changes (32). NOTE: Cancer derived cells and tissues also maintain short telomeres within a limited size range, mean TRF ~3-8 kb (26,27,86,91). This is shorter than what is seen in normal finite human cells. Thus, cancer-associated immortalization, encompassing conversion, appears to be a unique process, generating cells with unique telomere dynamics.
While we have gained information about the molecular properties associated with conversion, much about this process remains unknown. Our current speculation is that conversion may reflect a need to alter chromosome conformation at the telomeres when cells employ cancer-associated immortalization to transition from a finite state (no stable telomere length maintenance) to one where sufficient telomerase maintains the short stable telomeres. As well studied in yeast, immortal cells can have “counting” mechanisms to maintain telomeres within a limited size range (92). Since most human carcinoma cells, as well as our immortal HMEC lines, maintain telomeres within a short range, some type of “counting” mechanism likely is involved. Short stable telomeres are not seen in normal telomerase expressing human cells such as stem cells and lymphocytes (93), suggesting that active processes may be required for conversion to the distinct telomeric state seen in the in vitro immortalized and immortal cancer-derived cells. Functional p53 may present a partial barrier to the conversion process until very short telomeres provoke a structural change at the telomeric ends. We speculate that conversion may provoke widespread changes in gene expression, thus accounting for the closer resemblance of non-malignant immortal cell in vitro and in vivo to malignant immortal cells rather than to normal finite cells. Significantly, since the conversion process to immortality is unique to cells on the pathway to cancer, inhibition of this cancer-associated immortalization process could be a very valuable therapeutic target, if more experimental attention were paid to the human carcinogenesis process rather than “models” that don’t actually model.
hTERT immortalization: We, and others, have also immortalized finite HMEC by ectopic overexpression of the hTERT gene (73,88,94,95). Initial lines came from transducing hTERT into post-stasis post-selection HMEC grown in serum-free medium (184BTERT, 48RTERT, 161TERT); consequently, these immortalized lines contain the many cancer-associated and other aberrancies found in this p16(-) metaplastic post-stasis type. We and others were unable to immortalize pre-stasis HMEC grown in serum-free media with transduced hTERT, and only rare clonal immortalization occurred when hTERT was transduced into pre-stasis HMEC grown in MM (184FTERT) (88). Hypothesizing that high stress exposure (p16 expression) might prevent hTERT immortalization of pre-stasis HMEC, we transduced hTERT into passage 3 pre-stasis 184D grown in low-stress M87A, prior to p16 elevation. Nearly uniform immortalization was observed, generating the non-clonal line 184DTERT1 (73).
NOTE: TERT reactivation during in vivo malignant progression is not caused by ectopic introduction. The errors and mechanisms responsible for endogenous TERT reactivation (immortalization) are key to understanding human tumorigenesis (and its potential prevention), and they cannot be investigated or recapitulated in lines created by TERT transduction. Most important, we have seen that ectopic TERT overexpression does not model cancer-associated immortalization; it creates lines with properties not found in normal or abnormal HMEC in vivo. TRAP activity is extremely high [unpublished], compared to more modest levels in most in vitro immortalized and cancer-derived lines (27). Where examined, mean TRF length is much longer than the typical 2-8 kb range of immortalized and cancer-derived lines (26,27,86,88), indicating that these TERT-immortalized lines did not undergo conversion and therefore have telomere dynamics distinct from most carcinoma cells in vivo. Telomerase expression can enable cells to maintain growth in the presence of TGFβ (88); other signaling pathways are also affected by transduced TERT (96). 184DTERT did not express and was not inducible for p16 [unpublished]. hTERT-immortalized human cells, unlike cells with endogenous telomerase reactivation, may remain initially sensitive to OIS (39,46). Based on such data, I do not think that TERT-immortalized HMEC represent accurate models for normal, aberrant, or cancer (including cancer stem) HMEC in vivo; rather their expression of in vitro induced artifacts can potentially obscure understanding of in vivo biology. Since these immortalized lines are aberrant compared to normal HMEC (which are finite, with no or low TRAP activity, and with intact stasis, OIS, and replicative senescence tumor suppressor barriers), they cannot be accurate normal controls. This is particularly true since most finite HMEC immortalized by hTERT are the already highly aberrant post-selection post-stasis cells.
In general, we have seen that different methods of producing immortal HMEC, e.g., culture medium, oncogenic agents, can yield cell lines with significantly different phenotypes. The age of the specimen donor may also influence phenotype. Immortalized lines generated from younger women tend to express a phenotype most similar to the basal subtype of human breast cancers, while more luminal phenotypes can be seen in from older women (Figure 7) (59). The use of the cyclin D1/CDK2 construct to bypass stasis promoted a more luminal phenotype, even in a younger specimen (59), consistent with the in vivo observation that D1-overexpressing cancers tend to be more luminal (97). Immortalization of the post-selection post-stasis types may favor a metaplastic phenotype (67). Possibly, the prior prevalence of immortalized lines with only a basal or metaplastic phenotype was a consequence of using cells from mostly younger women combined with stressful culture medium that did not support growth of normal HMEC with luminal or progenitor properties. Additionally, in vivo human malignant progression is influenced by interactions with the microenvironment, which our current culture conditions do not recapitulate.
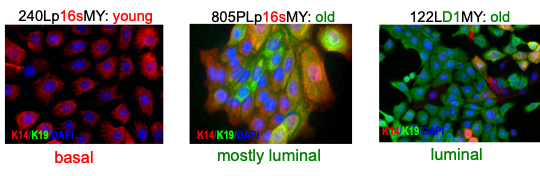
Figure 7. Phenotype of non-clonal immortal HMEC lines depends upon the age of the specimen donor and method to become post-stasis. Lineage specific keratin (K) protein expression shows that older women’s cells and overexpressed cyclin D1 bias to a luminal phenotype, while younger women’s cells and p16sh bias to basal (59).
Malignancy: Once the HMEC are immortally transformed and no longer vulnerable to OIS, the introduction of one or two oncogenes can further transform these cells towards malignancy (anchorage-independent growth, growth factor independence, and/or tumorigenicity in nude mice) (46,48-50). Finite lifespan HMEC cannot be rendered malignant by the same oncogenes because they are OIS-sensitive and die/senesce, underscoring the critical importance of the immortalization step in malignant progression. Thus, many reports in the literature show that one error can confer malignancy-associated properties to abnormal, immortally transformed lines such as MCF10A and 184B5 – because immortal lines have abrogated all the major tumor suppressor mechanisms! Unfortunately, the necessity for prior immortalization to achieve these results is commonly overlooked (particularly if the immortal lines are called normal or untransformed), sometimes giving the impression that one error can malignantly transform a normal HMEC (humans would all quickly die of cancer if this were true). Comparisons of non-malignant immortal lines with oncogene-exposed derivatives that had gained anchorage-independent growth did not show major differences in gene transcript profiling or global promoter methylation, in contrast to the major differences seen between all finite and all immortalized cultures (32,66,74). These data, along with the data on DCIS in vivo, are consistent with the acquisition of immortality, rather than the acquisition of malignancy, as the step in human carcinogenesis most associated with molecular alterations.
Altogether, these studies validate our model of the senescence barriers encountered by cultured HMEC, indicate that genomic instability per se is not needed for immortalization, and support the hypothesis that carcinoma-associated genomic instability (along with many “passenger” errors) may have its origin in the inherent instability induced by telomere dysfunction at replicative senescence. The reproducible ability to generate non-malignant (OIS resistant) immortalized lines lacking gross genomic errors can facilitate further investigation of the molecular underpinnings of the immortalization process and provides substrates to examine the effects of additional oncogenes and in-vivo identified genomic alterations on malignant progression. Most important, our studies highlight the critical and essential role of immortalization in carcinogenesis, and its potential as a therapeutic target. We believe the importance of the immortalization process is widely ignored because: 1) small rodents, lacking a significant immortalization barrier, do not model human carcinogenesis; 2) many scientists/journals incorrectly refer to non-malignant immortally transformed lines as “normal” or “untransformed”; 3) using hTERT to immortalize precludes study of actual in-vivo cancer-associated immortalization.
Concluding NOTE: Immortal HMEC have been actively transformed to immortality from their normal finite state. All normal human somatic cells are finite, and vulnerable to multiple tumor suppressor barriers (stasis, replicative senescence, OIS). The stringent replicative senescence barrier to immortality has evolved in large-long-lived animals like humans to suppress tumorigenesis. Immortality (expression of sufficient telomerase to maintain stable telomere lengths) is the most common alteration from normal associated with human solid cancers. We believe that attaining immortality is likely the most rate-limiting step in human carcinogenesis – immortally transformed lines such as 184A1 and MCF10A have acquired the errors that allowed them to overcome all tumor suppressor barriers, so that the overexpression of one oncogene can confer malignancy. Our immortal lines cluster with tumor derived lines and not finite HMEC in properties such as gene expression, promoter methylation, and resistance to OIS and TGFβ growth inhibition. Immortal lines may be non-malignant, but they are NOT normal, “normal”, or untransformed. Please do not refer to immortal HMEC (or any immortalized human cells) as normal or untransformed. If by “untransformed” you mean not malignantly transformed, and by “transformed” you mean malignant, then say so. Otherwise, this fosters the scientifically incorrect impression that immortality is a property of normal cells, rather than the reality that in humans it is only associated with pre-malignant and malignant somatic cells. This misrepresentation can impair development of preventive approaches. I sometimes despair about how we will be able to understand and develop therapeutics for early stage human epithelial carcinogenesis when immortal cell lines such as 184A1 and MCF10A, or TERT-immortalized post-selection post-stasis HMEC, are routinely referred to in the literature as normal or untransformed, employed as “normal” controls, or used as a starting point to study “early stage” carcinogenesis. I view this as similar to calling telomerase(+) DCIS “normal”, and using it as a normal control for cancer or the starting point for studying early stage carcinogenesis. There is already much in the literature to indicate that many, if not most of the significant alterations seen in breast and other carcinomas are already present in the pre-malignant stage; early stage progression is from normal finite to immortal DCIS.
References
72. Gollahon LS, Shay JW. Immortalization of human mammary epithelial cells transfected with mutant p53 (273his). Oncogene 1996;12:715-25.